|
Kommission Somatische Gentherapie des Wissenschaftlichen Beirats |
|
Die mit den Richtlinien zum Gentransfer in menschliche Körperzellen 1995 eingerichtete Kommission Somatische Gentherapie hat in Abstimmung mit dem Arbeitskreis Medizinischer Ethik-Kommissionen ein Regelungskonzept für klinische Studienvorhaben unter Verwendung von Gentransfer-Arzneimitteln etabliert. Kernaufgabe der Kommission ist die fachlich-inhaltliche Begutachtung dieser Studien, die der zuständigen lokalen Ethikkommission als Beratungsgrundlage für ihr nach dem Arzneimittelgesetz erforderliches Votum zur Verfügung gestellt wird. Im Jahr 2002 wurden 14 Anträge bzw. Amendments zur Begutachtung bei der Kommission vorgelegt. Von diesen wurden 5 befürwortet und eines abgelehnt. 6 Antragstellern wurden die Bedenken der Kommission im Sinne eines Zwischenvotums mit der Bitte um Überarbeitung des Studienprotokolls mitgeteilt. Ein laufendes Beurteilungsverfahren aus dem Vorjahr konnte mit einem positiven Votum abgeschlossen werden; in zwei weiteren Fällen aus dem Jahr 2001 wurde ein Zwischenvotum erstellt. Zusätzlich hat die Kommission eine Forschergruppe im Vorfeld der Antragsstellung ausführlich beraten. Die Charakteristika der im Jahr 2002 eingereichten Anträge sind in der Tabelle zusammengefasst. Unterbrechung klinischer Prüfungen unter Verwendung lebender, retroviral modifizierter Zellen Die Arbeit der Kommission war im Jahr 2002 maßgeblich bestimmt durch zwei schwere Verdachtsfälle unerwünschter Arzneimittelwirkungen, die im Rahmen einer französischen Gentherapiestudie zur Behandlung von Kindern, die an der erblichen Immunschwäche-Krankheit SCID-X1 (Typ X1 der Severe Combined immunodeficiency Disease) leiden. Innerhalb eines halben Jahres wurde im Rahmen dieser Studie über Leukämiefälle bzw. leukämie-ähnliche Erkrankungen bei zwei der behandelten Kindern berichtet. Zuvor war in der Literatur aufgrund neuerer tierexperimentelle Ergebnisse ein tumorigenes Potential für ein bestimmtes Markergen postuliert worden. Die Bewertung der experimentellen und klinischen Daten sowie die Konsequenzen hieraus auf in Deutschland laufende Studien mit retroviral modifizierten lebenden Zellen wurden in enger Abstimmung mit dem Paul- Ehrlich Institut jeweils zeitnah erörtert. Am 17.09.2002 wurde hierzu eine Anhörung und ein Erfahrungsaustausch unter Teilnahme internationaler Experten durchgeführt. Im Ergebnis wurden Empfehlungen an die zuständigen lokalen Ethikkommissionen zur temporären Aussetzung der positiven Voten zu allen Studien unter Verwendung lebender, retroviral modifizierter Zellen ausgesprochen. Nur zwei dieser Studien wurden nach weiteren Beratungen zur Nutzen-Risiko-Analyse zur Weiterführung nach Prüfplanänderung bzw. Modifikation der Patienteninformationen empfohlen; über eine Studie ist nach Protokolländerung erneut zu beraten; zwei neu eingereichte Protokolle werden im üblichen Verfahren beurteilt werden. Maßgebend für die Voten der Kommission war das Schutzinteresse der behandelten Patienten vor noch nicht abschließend beurteilbaren Risiken. Die Öffentlichkeit wurde in insgesamt vier gemeinsam mit dem Paul-Ehrlich-Institut herausgegeben Mitteilungen (http://www.ksg.baek.de und http://www.pei.de) über die jeweiligen Ereignisse und Konsequenzen für die Gentherapiestudien in Deutschland informiert. Die Zusammenarbeit mit den lokalen Ethikkommissionen funktionierte vorbildlich. Prüfkriterienkatalog zur Standardisierung der Antragsstellung Um eine Standardisierung der Antragsstellung bei der Kommission zu erreichen, hat eine Arbeitsgruppe der Kommission einen ausführlichen Prüfkriterienkatalog erstellt. Dieser Katalog ist im Internet (http://www.ksg.baek.de) verfügbar, damit sich insbesondere die Antragsteller bei der Ausarbeitung ihrer Studienprotokolle frühzeitig über die gestellten Anforderungen orientieren können. Zugleich soll durch die Offenlegung der Beurteilungskriterien auch die Transparenz der Antragsbegutachtung durch die Kommission für die gesamte Fachöffentlichkeit erhöht werden. GCP-Richtlinie 2001/20/EG vom 04.04.2001 Die Umsetzung der Richtlinie 2001/20/EG des Europäischen Parlaments und des Rates zur Angleichung der Rechts- und Verwaltungsvorschriften der Mitgliedstaaten über die Anwendung der guten klinischen Praxis bei der Durchführung von klinischen Prüfungen mit Humanarzneimitteln in deutsches Recht war Gegenstand mehrerer Beratungen. Dieser Prozess wird weiterhin intensiv zu begleiten sein, um auch zukünftig ein angemessenes Begutachtungssystem zum Schutz der Patienten vor spezifischen Risiken bei Studien mit Gentransfer sicherzustellen. Gentransferstudien-Register Die in Deutschland durchgeführten klinischen Studien mit Gentransfer werden über ein zentrales Register am Koordinierungszentrum für Klinische Studien (KKS) in Freiburg dem Deutschen Register für Somatische Gentransferstudien (DeReG) dokumentiert und evaluiert. Das KKS kooperiert hierzu mit der Kommission Somatische Gentherapie und der Deutschen Arbeitsgemeinschaft für Gentherapie (DAG-GT e.V.). Nach Ablauf der Konkretisierungsphase hat das BMBF im vergangenen Jahr den Antrag auf Weiterführung des Forschungsprojektes, das dem Aufbau des DeReG dient, positiv beschieden. Ein Ziel des Registers ist es, durch Bereitstellung von Informationen über abgeschlossene und laufende Studien Studienleiter bei der Planung und Durchführung von klinischen Studien zu unterstützen. Zwischenzeitlich wurde eine Internetseite eingerichtet, auf der auch in allgemeinverständlicher Form über Gentransferstudien berichtet wird sowie aggregierte Daten zu den in Deutschland durchgeführten Studien aufgeführt sind (http://www.zks.uni-freiburg.de/dereg.html). Derzeit wird eine benutzerfreundliche Oberfläche erstellt, die Patienten, Wissenschaftlern, interessierten Laien und Medienvertretern zur Verfügung stehen soll. 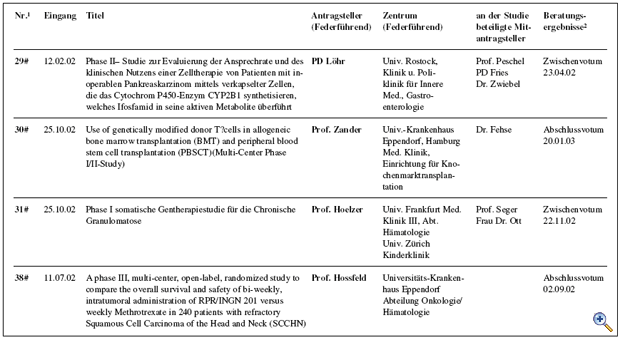 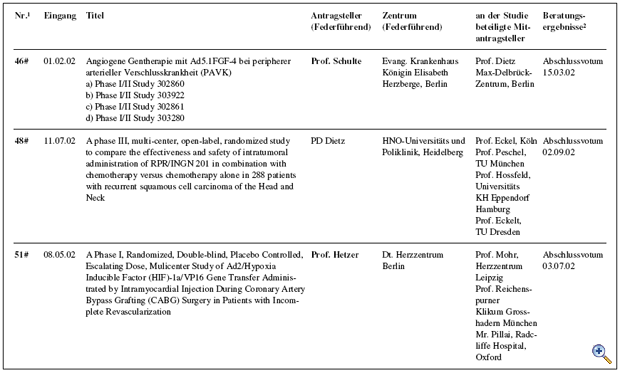 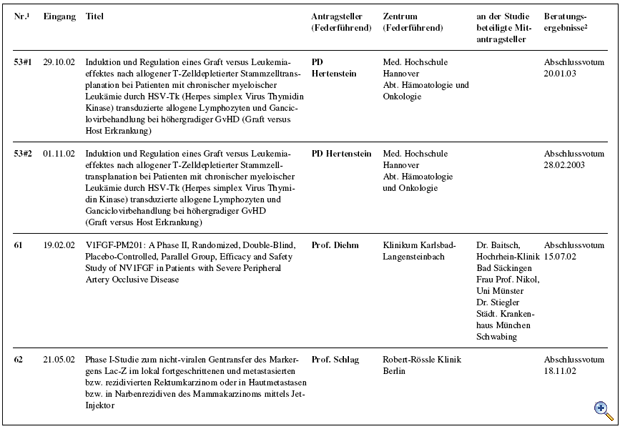 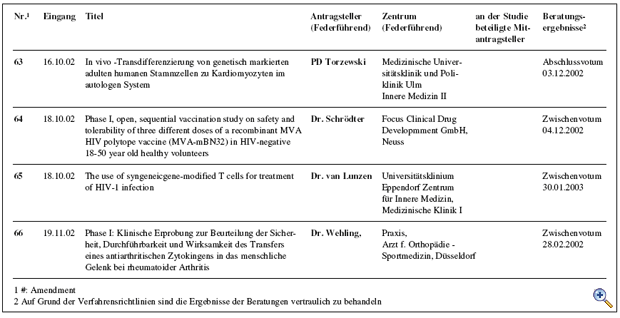
Abb.: Übersicht der Antragsteller, Zentren, beteiligte Mitantragsteller und Beratungsergebnisse |
| © 2003, Bundesärztekammer. |